One of the main objectives of our laboratory is to study the establishment of HIV latency coupled with stochastic HIV transcription and the host functional genome. Using quantitative gnomic and machine-learning-based approaches tailored from the laboratory, we aim to deepen our grasp on how variations, with resolution of single viruses, in HIV genome integrity and transcription influence the configuration of HIV reservoirs. Two research lines aligning with this objective are conducted.
1. Role of HIV antisense transcripts (AST) in the establishment of latency
Although the existence of HIV AST and encoded proteins was first postulated by computer analyses in 1988, at present the fundamental mechanisms behind HIV AST are not fully understood. HIV antisense RNAs have been demonstrated in vitro to be capable of promoting the initiation and maintenance of HIV latency; however, HIV AST in vivo is poorly detected and the observations are contradictory, leaving uncertainty to conclude the mechanism of how HIV AST contributes to its pathogenesis. We characterize the possible threshold of the ratio between HIV sense and antisense transcription, determining the transcriptional phenotypes of HIV based on barcoded HIV and cellular clones possessing unique transcriptional phenotypes of HIV.
2. Distinguishable topology of the task-evoked functional genome networks in HIV reservoirs
Our len to view HIV reservoir is that it can be represented as the topological (task-evoked) property of a network consisting of different communities of the genes targeted by HIV. Such communities are so-called immunologic signatures. HIV integration frequency within a network might be used as a proxy to define specific immune cell types and proinflammatory soluble factors, facilitating fine-tuning of the microenvironment of reservoirs.
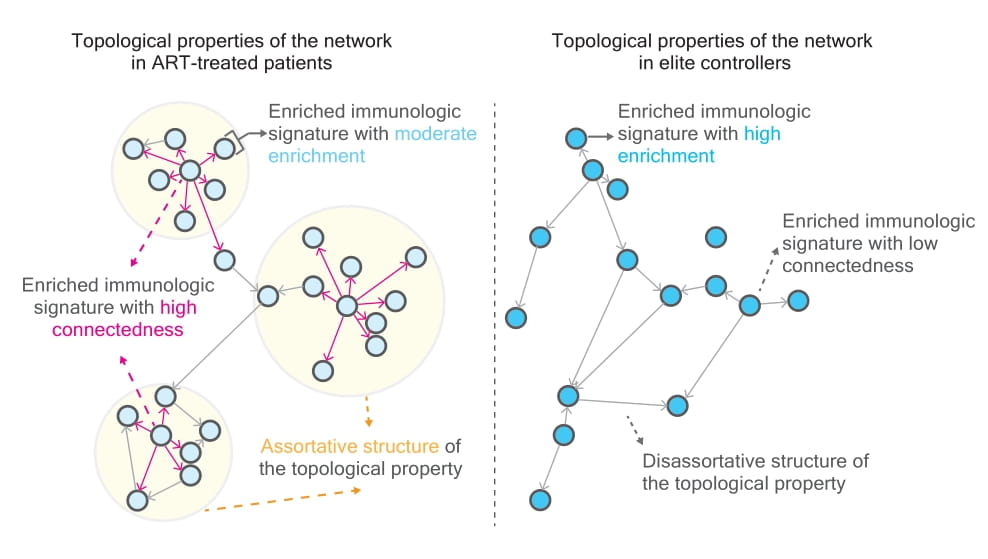
To deepen our understanding of such heterogeneous HIV reservoirs and their functional implications, we pioneer the integration of a convergence approach to characterize the composition of HIV reservoirs.Based on graph-theoretical tools, we observe noticeable topological properties in networks, featuring immunologic signatures enriched by genes harboring intact and defective proviruses, when comparing antiretroviral therapy (ART)-treated HIV-1-infected individuals and elite controllers.
The key variable, the rich factor, plays a pivotal role in classifying distinct topological properties in networks. The host gene expression strengthens the accuracy of classification between elite controllers and ART-treated patients. Markov chain Monte Carlo modeling for the simulation of different graph networks demonstrated the presence of an intrinsic barrier between elite controllers and non-elite controllers. Notably, our work provides a prime example of leveraging genomic approaches alongside mathematical tools to unravel the complexities of HIV reservoirs.
Figure 1. The network topology of HIV reservoirs in ART-treated patients versus elite controllers. In comparison to elite controllers, the network architecture in ART-treated patients exhibits three key characteristics (1) a less intense magnitude of signature enrichment, (2) a high degree of assortativity, and (3) elevated connectedness between two adjacent vertices. These findings suggest that the network architecture is more connective and structural in ART-treated patients.
In ourlaboratory, we alsostudyzoonoticviralinfection. Westudy the linkagebetween the intrinsictropisms of coronavirusvariants and host domestication.
3. Identification of potential SARS-CoV-2 genetic markers resulting from host domestication
We develop a k–mer–basedpipeline, namely the PathogenOriginRecognitionToolusingEnrichedK–mers (PORT-EK) to identifygenomic regions enriched in the respectivehostsafter the comparison of metagenomes of isolatesbetweentwo host species.
Using it we successfullyidentifythousands of k–mersenriched in US white-taileddeer and betacoronaviruseswhilecomparingthem with humanisolates. In addition, we demonstratedifferentcoveragelandscapes of k–mersenriched in deer and bats and unravel 144 mutations in enrichedk–mersyielded from the comparison of viralmetagenomesbetween bat and humanisolates.
Additionally, we observethat the third positionwithin a geneticcodonisprone to mutations, resulting in a high frequency of synonymousmutations of aminoacidsharboring the same physicochemicalproperties as unalteredaminoacids. Importantly, we areable toclassify and predict the likelihood of host speciesbased on the enrichedk-mer counts.
Figure 2. Rational design of PORT-EK and determination of the enriched k-mers. (A) The analytical pipeline of PORT-EK. PORT-EK consists of four steps including (1) k-mers matrix preparation, (2) k-mers filtering and selection, (3) the identification of host-specific mutations, and (4) the classification of hosts. Details are described in the main text. (B) Funnel plot representing the filtering strategies for the selection of enriched k-mers. Four layers of filtering are applied in the PORT-EK pipeline.